챕터 6 - 와일드카드로 규칙 일반화하기
sketch_genomes_ 규칙 중 하나를 다시 살펴봅시다:
rule sketch_genomes_1:
input:
"genomes/GCF_000017325.1.fna.gz",
output:
"GCF_000017325.1.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""여기에는 중복이 있습니다. 접근 번호 GCF_000017325.1이 두 번 등장합니다. 이것을 개선할 수 있을까요?
가능합니다! Snakemake의 와일드카드(wildcards) 기능을 사용하면 Snakemake가 자동으로 채워 넣을 빈칸을 만들 수 있습니다.
와일드카드를 사용하면, {와 }로 와일드카드 이름을 감싸서 입력 또는 출력 파일 이름의 특정 부분을 대체 가능한 빈칸으로 표시할 수 있습니다. accession이라는 와일드카드를 만들고 규칙의 input: 및 output: 블록에 넣어봅시다:
rule sketch_genomes_1:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""이것이 의미하는 바는, .fna.gz.sig로 끝나는 출력 파일이 필요할 때마다 그 앞부분(.fna.gz.sig 이전의 텍스트)을 genomes/ 디렉토리에서 .fna.gz로 끝나는 파일을 찾아 존재하면 해당 파일을 입력으로 사용하라는 것입니다.
(네, 규칙에 와일드카드가 여러 개 있을 수 있습니다! 나중에 보여드리겠습니다!)
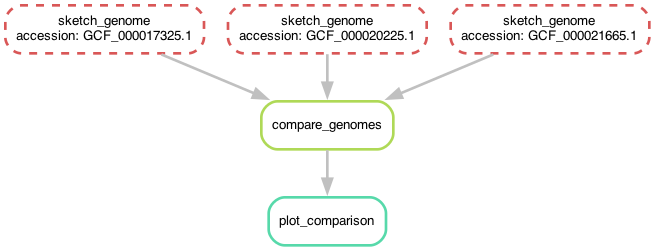
sketch_genomes_2와 sketch_genomes_3에도 와일드카드를 사용하면, 세 규칙이 완전히 동일해짐을 알 수 있습니다. 그리고 실제로 규칙은 하나만 있으면 됩니다 (사실 하나만 있을 수 있습니다). 이제 세 규칙을 다시 하나의 sketch_genome 규칙으로 통합할 수 있습니다.
전체 Snakefile은 다음과 같습니다:
rule sketch_genome:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
rule compare_genomes:
input:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
output:
"compare.mat"
shell: """
sourmash compare {input} -o {output}
"""
rule plot_comparison:
message: "sourmash로 모든 입력 게놈을 비교합니다"
input:
"compare.mat"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot {input}
"""처음 시작했던 Snakefile과 매우 비슷해 보이지만, 핵심적인 차이점은 이제 와일드카드를 사용한다는 것입니다.
이전 섹션 끝에서의 상황(세 게놈을 한꺼번에 스케치하는 한 규칙)과 달리, 이제는 한 번에 게놈 하나씩 스케치하지만 병렬로도 실행할 수 있는 규칙이 생겼습니다! 따라서 snakemake -j 3은 여전히 작동하며, 게놈을 더 추가하거나 동시 실행 작업 수를 늘려도 계속 잘 동작할 것입니다.
이 작업을 계속하기 전에, 워크플로가 어떤 모양인지 다시 살펴봅시다. 모양은 같지만 약간 달라 보일 것입니다! 이제 게놈 스케치 규칙들은 서로 다른 이름 대신 모두 같은 이름을 가지지만, accession 와일드카드에 서로 다른 값을 가집니다!