챕터 1 - Snakemake가 프로그램을 대신 실행해 줍니다!
생물정보학 분야에서는 시퀀싱 데이터를 분석하고 처리하기 위해 수많은 프로그램을 실행해야 하는 경우가 많습니다. Snakemake는 이 과정을 도와주는 도구입니다.
첫 번째, 간단한 Snakemake 워크플로
다음은 간단하면서도 실용적인 Snakemake 워크플로입니다:
rule compare_genomes:
message: "sourmash로 모든 입력 게놈을 비교합니다"
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
sourmash plot compare.mat
"""이 내용을 Snakefile이라는 파일에 저장하고 snakemake -j 1로 실행하세요.
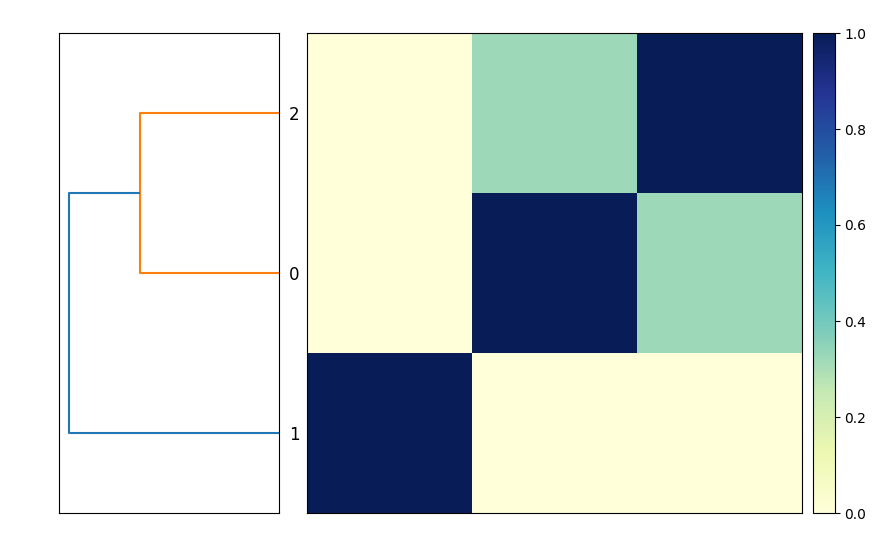
실행이 완료되면 세 게놈의 유사도 행렬과 덴드로그램이 담긴 출력 파일 compare.mat.matrix.png가 생성됩니다(그림 1 참고).
이것은 아래의 세 명령어를 compare-genomes.sh라는 파일에 넣고 bash compare-genomes.sh로 실행하는 것과 기능적으로 동일합니다:
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
sourmash plot compare.mat Snakemake 버전이 셸 스크립트보다 이미 더 편리한 이유는, 명령이 성공하면 초록색 텍스트로 “1 of 1 steps (100%) done!”이라는 메시지를, 실패하면 빨간색 텍스트로 알림을 보여주기 때문입니다.
하지만 Snakemake 버전을 셸 스크립트 버전보다 훨씬 더 개선할 수 있습니다!
불필요한 명령 재실행 방지: 두 번째 워크플로
snakemake -j 1을 실행할 때마다 명령들이 다시 실행됩니다. 하지만 이미 원하는 출력 파일이 있다면 굳이 다시 실행할 필요가 없습니다!
어떻게 하면 Snakemake가 규칙을 다시 실행하지 않도록 할 수 있을까요?
shell: 블록 앞에 output: 블록을 추가해 Snakemake에게 예상 출력 파일을 알려주면 됩니다:
rule compare_genomes:
message: "compare all input genomes using sourmash"
+ output:
+ "compare.mat.matrix.png"
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first이제 snakemake -j 1을 처음 실행하면 명령들이 실행되지만, 두 번째로 실행하면 “Nothing to be done (all requested files are present and up to date).”라고 표시됩니다.
위의 코드 예제는 다소 낯설어 보일 수 있습니다. 앞에 ’+’가 붙은 줄이 있고 초록색으로 표시되어 있습니다. 이게 무엇일까요?
이것은 diff 프로그램이 생성한 두 소스 코드 파일의 줄별 비교인 “diff”입니다. 여기서 diff는 원본 코드 목록에 두 줄이 추가되었음을 보여줍니다 (‘+’ 로 시작하는 두 줄이 추가된 줄입니다). 또한 추가된 줄 위아래에 문맥을 제공하여 원본 코드의 어디에 추가되었는지 쉽게 파악할 수 있습니다.
아래에서는 제거된 줄의 예시도 볼 수 있는데, 첫 번째 위치에 ’-’가 표시되고 빨간색으로 강조됩니다.
원하는 출력 파일 compare.mat.matrix.png가 이미 존재하기 때문에 Snakemake는 아무것도 할 필요가 없다는 것을 알 수 있습니다!
compare.mat.matrix.png를 삭제하고 snakemake -j 1을 다시 실행하면, Snakemake가 파일을 다시 만들어 줍니다:
$ rm compare.mat.matrix.png
$ snakemake -j 1이처럼 Snakemake를 사용하면 이미 원하는 파일이 있을 때 명령 재실행을 손쉽게 피할 수 있습니다. 셸 스크립트는 이런 기능을 자동으로 제공하지 않기 때문에, 생물정보학 워크플로에 Snakemake 같은 워크플로우 시스템을 사용하는 주요 이유 중 하나입니다.
필요한 명령만 실행하기
위의 마지막 Snakefile에는 세 개의 명령이 있지만, compare.mat.matrix.png만 삭제한다면 마지막 명령만 다시 실행하면 됩니다. 앞의 두 명령으로 생성된 파일은 이미 존재하므로 다시 만들 필요가 없습니다. 하지만 Snakemake는 그것을 알 방법이 없습니다. 전체 규칙을 하나의 단위로 처리하며, 셸 명령 내부를 들여다보며 무엇을 실행할 필요가 없는지 파악하지 않습니다.
이미 존재하는 파일을 다시 만드는 것을 피하려면 Snakefile을 조금 더 복잡하게 만들어야 합니다.
먼저, 명령들을 세 개의 별도 규칙으로 분리해 봅시다.
rule sketch_genomes:
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
shell: """
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
"""
rule plot_comparison:
message: "sourmash로 모든 입력 게놈을 비교합니다"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot compare.mat
"""그다지 복잡한 것은 없습니다. 두 개의 새로운 규칙 블록을 만들고, 각 규칙에 이름을 붙이고, 셸 명령을 나누어 각각의 규칙 블록에 배치했을 뿐입니다.
세 규칙 모두 실행하도록 Snakemake에 요청할 수 있습니다:
snakemake -j 1 sketch_genomes compare_genomes plot_comparison모두 성공적으로 실행됩니다!
하지만 여전히 일부 명령들이 매번 다시 실행되는 문제가 있습니다. compare.mat.matrix.png가 존재하기 때문에 plot_comparison은 매번 실행되지 않지만, sketch_genomes와 compare_genomes는 반복해서 실행됩니다.
어떻게 고칠 수 있을까요?
각 규칙에 output 블록 추가하기
각 규칙에 output: 블록을 추가하면, Snakemake는 출력 파일이 갱신되어야 하는 (예를 들어, 파일이 존재하지 않는) 규칙만 실행합니다.
적용해 봅시다:
rule sketch_genomes:
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p k=31 genomes/*.fna.gz --name-from-first
"""
rule compare_genomes:
output:
"compare.mat"
shell: """
sourmash compare GCF_000021665.1.fna.gz.sig \
GCF_000017325.1.fna.gz.sig GCF_000020225.1.fna.gz.sig \
-o compare.mat
"""
rule plot_comparison:
message: "sourmash로 모든 입력 게놈을 비교합니다"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot compare.mat
"""이제
snakemake -j 1 sketch_genomes compare_genomes plot_comparison를 실행하면, 출력 파일이 그대로 있는 한 각 명령이 딱 한 번씩만 실행됩니다. 만세!
하지만 여전히 세 규칙의 이름을 올바른 순서로 모두 지정해야 합니다. 번거롭습니다! 다음에 이것을 해결해 봅시다.